Since the discovery of the so-called “glucose-fatty acid cycle” in 1963, there has been more and more evidence accumulating linking free fatty acids with diabetes (Randle, Garland, Hales, & Newsholme, 1963). Briefly, the glucose-fatty acid cycle describes a competition, whereby the use of glucose becomes impaired by the presence of fatty acids and, to a lesser extent, vice versa (Cook, King, & Veech, 1978).
Others had hinted at this fatty acid-induced blocking effect of sorts before. Among them, was a guy named Apollinaire Bouchardat, a French pharmacist, who gave his diabetic patients sweet fruit and bread made from gluten, and had good results. Others had applied Bouchardat’s dietary prescription and achieved equally good success. More recently, an English physician, William Budd, following on the heels of these pioneers, gave his diabetic patients around 8 ounces of sugar daily, with only the intent of slowing the cachexia that was characteristic of long-standing poorly controlled diabetes. Not only did most of his patients stop wasting away, but they also began to stop losing sugar in their urine when given this “saccharine treatment” (Hughes, 1862).
Another prominent clinician-researcher, Harold Himsworth, who was also first to show that insulin sensitivity in the tissues is reduced in diabetics, decades later, suggested, based on his clinical experiences and a review of the population data that high intakes of dietary fat (which raises free fatty acid levels) caused diabetes, and that diets rich in carbohydrates and low in fat were protective of it (Himsworth, 1934a, 1934b, 1936).
Kahn et al. thereafter, via multivariate analyses, in effect, reconfirmed the data that had been amassed by Himsworth, by showing that men who ate more sugar were less likely to develop diabetes later on than those who ate less of it—believe it or not (Kahn et al., 1971). By the 1980s, sugar had been pretty much exonerated from the diseases that were, speculatively, being assigned to it.
The experiences of these researchers are at odds with the conception firmly held by most contemporary clinicians and lay people, that diabetes is a disorder of having too much glucose or insulin in the blood. If this were the case, restricting carbohydrates or taking a drug that lowers blood glucose or insulin levels to normal, should be expected to change the course of diabetes 180 degrees.
The results of three relatively recent clinical trials, though far from perfect, softened the confidence in this approach. Briefly in the trials, older diabetics were assigned to receive either the standard of care or a more intensive regimen, in which blood glucose levels, by various means, were to be lowered to normal, or near-normal, levels for the purpose of reducing deaths from strokes and heart attacks. Taken together, more deaths were incurred from all causes in the subjects whose blood glucose levels were reduced to normality the most (Gerstein et al., 2008; Patel et al., 2008; Duckworth et al., 2009).
(The so-called “insulin-sensitizer” rosiglitazone immediately comes to mind, which now bears a black box warning about this increased cardiovascular risk.)
This begs the question, if carbohydrates and sugar are not the cause of diabetes, and if elevated levels of glucose and insulin aren’t either, ideas that have been bandied about among bloggers and very popular health- and diet-websites, what exactly is? Bearing in mind the incipient defect in diabetes—that is, insulin resistance—the glucose-fatty acid cycle that was previously described, and assimilating the clinical experiences of Bouchardat and others, we will first and foremost, turn to the enzyme complex pyruvate dehydrogenase (PDH) in exploring this question.
The PDH complex is a crucial enzyme that links the metabolism of glucose via glycolysis in the cytoplasm and via the Krebs cycle in the mitochondrion. I’ve discussed elsewhere that the major inhibition of glucose is taking place here, rather than at the shuttling of glucose into the cell through the plasma membrane, for instance (Randle & Smith, 1958a, 1958b).
Fatty acids, both directly and indirectly, inactivate the PDH complex, preventing the metabolism of glucose to carbon dioxide, whereas carbohydrates have the opposite effect. Diabetics generally have elevated levels of free fatty acids in the blood, and this accounts for many processes that go off the rails in them, including (1) an impaired ability to switch from one fuel source (e.g., fat) to another (e.g., glucose) and (2) a 2- to 4-fold increased risk of cardiovascular disease compared to non-diabetics.
The inhibition of the PDH complex prevents the metabolism of pyruvate to acetyl CoA, the point at which the metabolism of carbohydrate, fat, and protein converge (figure 1).
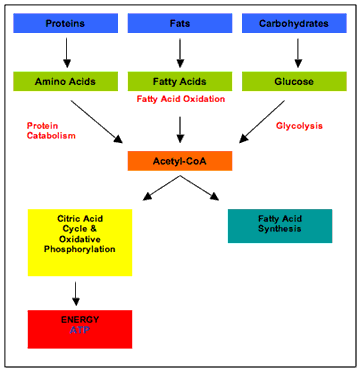 |
Figure 1 Overview of metabolism This in turn, backlogs the glycolytic enzymes upstream of the PDH complex. As as a result, free glucose molecules accumulate in the cell, and following their concentration gradient, move back out into the bloodstream (Sonksen, 2001). PDH is not only inhibited by fatty acids, but also by the products of their oxidation (Bowker-Kinley, Davis, Wu, Harris, & Popov, 1998). Conversely, glucose oxidation supplies pyruvate, which directly stimulates PDH activity, and activates (and increases the expression of) acetyl CoA carboxylase, an enzyme that puts a brake on fatty acid oxidation (Bouzakri et al., 2008) by way of generating malonyl CoA (figure 2). This latter process is referred to as a carboxylation reaction and as such, carbon dioxide, which is generated in greater amounts by glucose oxidation than fatty acid oxidation, pushes the reaction forward. |
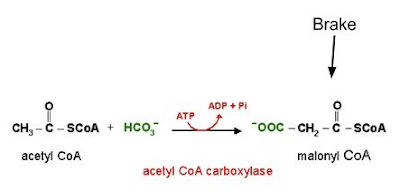 Figure 2 Malonyl CoA , generated from glucose oxidation, puts a "brake" on fatty acid oxidation. |
PDH, which is highly regulated, is neither altered nor transcribed less abundantly. Instead, PDH is more likely being kept in an inactive state by various factors, namely the free fatty acids, ketone bodies, and lactate—the levels of which are elevated in diabetes, in accordance with the original experimental observations that led to the discovery o the glucose-fatty acid cycle (McCromack, Edgell, & Denton, 1982).
Though both fatty acids and glucose generate acetyl CoA, the similarities stop there.
Though both fatty acids and glucose generate acetyl CoA, the similarities stop there.
For one, glucose oxidation reduces the oxygen costs of energy (ATP) generation, and ATP regulates, through mechanisms that are more speculative, the passage of glucose into cells. The oxidation of fatty acids consumes more oxygen than the oxidation of glucose does, and so leads to a lower oxygen tension therein.
The reduction in the oxygen tension in turn, stimulates the proliferation of fibroblasts that secrete collagen, and this newly laid collagen reduces the oxygen tension further by extending the distance by which oxygen has to diffuse to reach cells. The fibroblasts thereafter lay down more collagen and so on, in a typical positive feedback fashion, and this could eventually lead to greater degrees of atherosclerosis, as cholesterol, compared to other lipids, requires the greatest amount of oxygen for its oxidation.
Drugs that shift the preferential fuel use from fatty acids to glucose (some of which are in the pipeline) have been shown to be highly protective for diabetics and as a side effect, cardiovascular disease. This is because ATP is generated with a greater oxygen economy. Metabolic stress, which is elicited by the rapid depletion of ATP, can override the glucose-fatty acid cycle, in which case glucose would be converted to lactate rather than carbon dioxide (Bergeron et al., 1999; Young, Radda, & Leighton, 1996).
On the other hand, the currently used oral “hypoglycemic” drugs carry the unwanted side-effect of death from strokes and heart attacks (uncovered first in the 1970s by the University Research Diabetes Group)—events for which the task of lowering blood glucose levels was undertaken in the first place. (The evidence that these drugs [not insulin] protect against cardiovascular disease is also sparse.)
Two, the excessive delivery of fatty acids to tissues, as a protective reflex of sorts, increases the expression of uncoupling proteins therein, leading to a decreased generation of ATP (and the subsequent impaired functioning of tissues). In the pancreas, for instance, the increased expression of the uncoupling proteins, by depleting ATP, keeps the voltage-gated calcium channels closed, the opening of which is required for the secretion of insulin (figure 3) (Zhang et al., 2001).
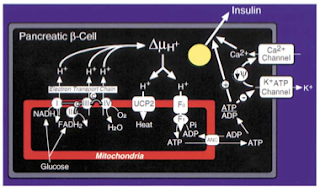 |
Figure 3 The uncoupling proteins (UCP) are the means by which the electrons derived from glucose are dissipated as heat, rather than employed to generated ATP. ATP is needed to stimulate insulin secretion from the beta cells of the pancreas (Zhang et al, 2001). |
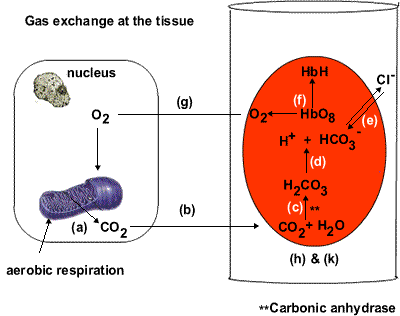
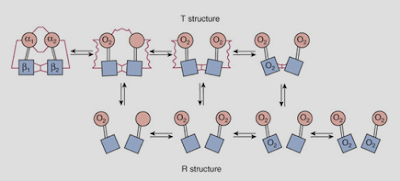
Three, glucose oxidation, more than fatty acid oxidation, leads to a greater production of gas, carbon dioxide. The unloading of oxygen from red blood cells to tissues is blunted as a result (per the Bohr effect), further reducing the oxygen levels (figure 4). (This is reflected in the respiratory quotient.) Diabetics, as a rule, expire less carbon dioxide than non-diabetics do (Simonson, Tappy, Jequier, Felber, & DeFronzo, 1988).
.gif)
.png) |
Figure 4 Top: The deoxygenated state of hemoglobin is maintained by the cell's acidity (H+) namely through the production of carbon dioxide (CO2), which carboxylates the amine (NH2) groups on the hemoglobin proteins (not shown), enhancing their acidity, and so their ability to form salt linages with other positively-charged parts of the hemoglobin molecule. Hemoglobin's oxygen molecules are released to tissues as a result. Bottom: The salt linkages (red squiggly lines) stabilize the deoxygenated state of hemoglobin (T structure). The loss of salt linkages allows hemoglobin to bind oxygen molecules with a greater affinity (R structure). Four, a consequence inherent in a high rate of fatty acid oxidation is the buildup of fatty acid metabolites in the cell (such as ceramides and diacylglycerols), which ultimately impair the so-called “insulin receptor” via pathways that converge on the activation of protein kinase C (Erion & Shulman, 2010). |
Five, a lower respiratory quotient goes along with a lower cellular energy charge, and this alone could explain many processes that go off the rails in diabetes. For instance, when the energy charge in the cell decreases, sodium moves into the cell (reducing sodium levels in the blood) and the cell’s ability to retain potassium diminishes. This could account for why diabetics have higher-than-average levels of aldosterone, the secretion of which, from the adrenal cortices, is triggered by falling sodium, or rising potassium, concentrations (figure 5) (Hollenberg et al., 2004; Kraus, Jäger, Meier, Fasshauer, & Klein, 2005; Maalouf, Cameron, Moe, & Sakhaee, 2010).
 |
Figure 5 Regulation of aldosterone (Aldo) secretion by potassium (K+) and sodium (Na+) levels in the blood. |
Aldosterone is linked to many adverse consequences, including inflammation, vascular problems, loss in insulin sensitivity, and the replacement of healthy tissue with scar tissue. Lo and behold, drugs that antagonize the aldosterone (mineralocorticoid) receptors have been shown to slow the progression of diabetic complications (Bender, McGraw, Jaffe, & Sowers, 2013). The effects of aldosterone are at odds with those of insulin, in that insulin has a pro-metabolic effect, whereas aldosterone has an anti-metabolic effect.
Six, fatty acid oxidation increases the tendency for lactate and protons to accumulate, both of which are elevated in parallel with free fatty acids, and in obesity and diabetes (Chen, Varasteh, & Reaven, 1993; Reaven, Hollenbeck, Jeng, Wu, & Chen, 1988). Lactate, like free fatty acids, interferes with insulin signaling, reinforcing the insulin resistant state (Choi et al., 2002; Depré, Veitch, & Hue, 1993).
And seven, it’s conceivable that the excessive oxidation of fatty acids, especially against the backdrop of oxidative stress, could impair the functioning of tissues further through the generation of advanced glycation end (AGE) products from acetone (namely through its conversion to methylglyoxal) produced as a result of high rates of fatty acid oxidation in the liver. (Credit goes to Dr. Chris Masterjohn for providing clarification on this for me.) Hyperglycemia, it turns out, could assume a secondary, or permissive, role in this process.
So in summary, it’s my contention that the excessive mobilization and oxidation of fatty acids—a signature of sorts of the diabetic metabolism— impairs insulin actions, primarily by way of inhibiting the PDH complex (Koves et al., 2005). Simply put, when free fatty acid levels are elevated, the metabolism of glucose to carbon dioxide becomes impaired, and glucose is rerouted to lactate instead. This has important implications for the cardiovascular disease—of which, to recap, diabetics have an increased risk of.
So, in my roundabout way, how does this translate to recommendations that you could start applying now if you are diabetic or trending towards it? This isn’t my area of expertise and, really, others elsewhere have done a good job in this regard.
Nonetheless, tentatively, I would recommend, in no particular order, to (1) supplement with vitamin B1 (cofactor of the PDH complex) and (2) magnesium (helps retain ATP in the cell), (3) eat sugar as in fruit in preference to starches as in grains and tubers, (3) reduce excess body fat if you have it, and replace it with muscle, which serve as sinks for free fatty acids, (4) reduce the fat in the diet and replace them with carbohydrates and protein, keeping calories more or less the same. (I’ve found, and most dieters knows, that the loss of muscle occurs long before the fat stores become depleted, which is why carbohydrates are superior to fat during periods of weight loss, as carbohydrates are strongly anabolic), (5) opt for small meals over large ones in order to maintain steadier blood glucose levels over the course of a day, and (6) de-stress as much as possible by, for instance, getting into the habit of creating and writing down plans for how you will get an A on an upcoming exam, cope with a major life change, complete a paper, win an argument, etc. This can be more powerful than executing the plan itself . . . it has been for me at least.
REFERENCES
1. Randle, P. J., Garland, P. B., Hales, C. N. & Newsholme, E. A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–9 (1963).
2. Cook, G. A., King, M. T. & Veech, R. L. Ketogenesis and malonyl coenzyme A content of isolated rat hepatocytes. The Journal of biological chemistry 253, 2529–31 (1978).
3. Hughes, J. Quarterly summary of the improvements and discovereis in the medical sciences. The American Journal of the Medical Sciences 44, 232–235 (1862).
4. Himsworth, H. P. Dietetic factors influencing the glucose tolerance and the activity of insulin. The Journal of physiology 81, 29–48 (1934).
5. Himsworth, H. P. Management of diabetes mellitus. British medical journal 2, 137–41 (1936).
6. Himsworth, H. P. High carbohydrate diets and insulin efficiency. British medical journal 2, 57–60 (1934).
7. Kahn, H. A. et al. Factors related to diabetes incidence: a multivariate analysis of two years observation on 10,000 men. The Israel Ischemic Heart Disease Study. Journal of chronic diseases 23, 617–29 (1971).
8. Gerstein, H. C. et al. Effects of intensive glucose lowering in type 2 diabetes. The New England journal of medicine 358, 2545–59 (2008).
9. Patel, A. et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. The New England journal of medicine 358, 2560–72 (2008).
10. Duckworth, W. et al. Glucose control and vascular complications in veterans with type 2 diabetes. The New England journal of medicine 360, 129–39 (2009).
11. Randle, P. J. & Smith, G. H. Regulation of glucose uptake by muscle. 1. The effects of insulin, anaerobiosis and cell poisons on the uptake of glucose and release of potassium by isolated rat diaphragm. The Biochemical journal 70, 490–500 (1958).
12. Randle, P. J. & Smith, G. H. Regulation of glucose uptake by muscle. 2. The effects of insulin, anaerobiosis and cell poisons on the penetration of isolated rat diaphragm by sugars. The Biochemical journal 70, 501–8 (1958).
13. Sonksen, P. H. Insulin, growth hormone and sport. The Journal of endocrinology 170, 13–25 (2001).
14. Bowker-Kinley, M. M., Davis, W. I., Wu, P., Harris, R. A. & Popov, K. M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. The Biochemical journal 329 ( Pt 1, 191–6 (1998).
15. Bouzakri, K. et al. Malonyl CoenzymeA decarboxylase regulates lipid and glucose metabolism in human skeletal muscle. Diabetes 57, 1508–16 (2008).
16. McCormack, J. G., Edgell, N. J. & Denton, R. M. Studies on the interactions of Ca2+ and pyruvate in the regulation of rat heart pyruvate dehydrogenase activity. Effects of starvation and diabetes. The Biochemical journal 202, 419–27 (1982).
17. Bergeron, R. et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. The American journal of physiology 276, E938–44 (1999).
18. Young, M. E., Radda, G. K. & Leighton, B. Activation of glycogen phosphorylase and glycogenolysis in rat skeletal muscle by AICAR--an activator of AMP-activated protein kinase. FEBS letters 382, 43–7 (1996).
19. Chan, C. B. et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes 50, 1302–10 (2001).
20. Zhang, C. Y. et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745–55 (2001).
21. Simonson, D. C., Tappy, L., Jequier, E., Felber, J. P. & DeFronzo, R. A. Normalization of carbohydrate-induced thermogenesis by fructose in insulin-resistant states. The American journal of physiology 254, E201–7 (1988).
22. Erion, D. M. & Shulman, G. I. Diacylglycerol-mediated insulin resistance. Nature medicine 16, 400–2 (2010).
23. Hollenberg, N. K. et al. Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney international 65, 1435–9 (2004).
24. Kraus, D., Jäger, J., Meier, B., Fasshauer, M. & Klein, J. Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et métabolisme 37, 455–9 (2005).
25. Maalouf, N. M., Cameron, M. A., Moe, O. W. & Sakhaee, K. Metabolic basis for low urine pH in type 2 diabetes. Clinical journal of the American Society of Nephrology : CJASN 5, 1277–81 (2010).
26. Bender, S. B., McGraw, A. P., Jaffe, I. Z. & Sowers, J. R. Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes 62, 313–9 (2013).
27. Chen, Y. D., Varasteh, B. B. & Reaven, G. M. Plasma lactate concentration in obesity and type 2 diabetes. Diabète & métabolisme 19, 348–54 (1993).
28. Reaven, G. M., Hollenbeck, C., Jeng, C. Y., Wu, M. S. & Chen, Y. D. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37, 1020–4 (1988).
29. Depré, C., Veitch, K. & Hue, L. Role of fructose 2,6-bisphosphate in the control of glycolysis. Stimulation of glycogen synthesis by lactate in the isolated working rat heart. Acta cardiologica 48, 147–64 (1993).
30. Choi, C. S. et al. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. American journal of physiology. Endocrinology and metabolism 283, E233–40 (2002).
31. Koves, T. R. et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. The Journal of biological chemistry 280, 33588–98 (2005).